Description: Homo sapiens myosin VIIA (MYO7A), transcript variant 1, mRNA. RefSeq Summary (NM_000260): This gene is a member of the myosin gene family. Myosins are mechanochemical proteins characterized by the presence of a motor domain, an actin-binding domain, a neck domain that interacts with other proteins, and a tail domain that serves as an anchor. This gene encodes an unconventional myosin with a very short tail. Defects in this gene are associated with the mouse shaker-1 phenotype and the human Usher syndrome 1B which are characterized by deafness, reduced vestibular function, and (in human) retinal degeneration. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2008]. Transcript (Including UTRs) Position: hg19 chr11:76,839,310-76,926,286 Size: 86,977 Total Exon Count: 49 Strand: + Coding Region Position: hg19 chr11:76,841,681-76,925,741 Size: 84,061 Coding Exon Count: 48
ID:MYO7A_HUMAN DESCRIPTION: RecName: Full=Unconventional myosin-VIIa; FUNCTION: Myosins are actin-based motor molecules with ATPase activity. Unconventional myosins serve in intracellular movements. Their highly divergent tails bind to membranous compartments, which are then moved relative to actin filaments. In the retina, plays an important role in the renewal of the outer photoreceptor disks. Plays an important role in the distribution and migration of retinal pigment epithelial (RPE) melanosomes and phagosomes, and in the regulation of opsin transport in retinal photoreceptors. In the inner ear, plays an important role in differentiation, morphogenesis and organization of cochlear hair cell bundles. Involved in hair-cell vesicle trafficking of aminoglycosides, which are known to induce ototoxicity (By similarity). Motor protein that is a part of the functional network formed by USH1C, USH1G, CDH23 and MYO7A that mediates mechanotransduction in cochlear hair cells. Required for normal hearing. ENZYME REGULATION: ATP hydrolysis is inhibited by Mg(2+), already at a concentration of 0.4 mM. SUBUNIT: Interacts with PLEKHB1 (via PH domain). Interacts with PCDH15. Interacts with RPE65. Interacts with TWF2 (By similarity). Might homodimerize in a two headed molecule through the formation of a coiled-coil rod. May interact with CALM. Binds MYRIP and WHRN. Identified in a complex with USH1C and USH1G. SUBCELLULAR LOCATION: Cytoplasm. Cytoplasm, cell cortex. Cytoplasm, cytoskeleton. Note=In the photoreceptor cells, mainly localized in the inner and base of outer segments as well as in the synaptic ending region. Colocalizes with a subset of melanosomes in retinal pigment epithelium cells. Detected at the tip of cochlear hair cell stereocilia. The complex formed by MYO7A, USH1C and USH1G colocalizes with F-actin. TISSUE SPECIFICITY: Expressed in the pigment epithelium and the photoreceptor cells of the retina. Also found in kidney, liver, testis, cochlea, lymphocytes. Not expressed in brain. DEVELOPMENTAL STAGE: Detected in optic cup in 5.5 weeks-old embryos. Expressed in retinal pigment epithelium, cochlear and vestibular neuroepithelia, and olfactory epithelium at 8 weeks. At 19 weeks, present in both pigment epithelium and photoreceptor cells. At 24-28 weeks, expression in pigment epithelium and photoreceptor cells increases. Present in pigment epithelium and photoreceptor cells in adult. DISEASE: Defects in MYO7A are the cause of Usher syndrome type 1B (USH1B) [MIM:276900]. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa and sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH1 is characterized by profound congenital sensorineural deafness, absent vestibular function and prepubertal onset of progressive retinitis pigmentosa leading to blindness. DISEASE: Defects in MYO7A are the cause of deafness autosomal recessive type 2 (DFNB2) [MIM:600060]; also called neurosensory non-syndromic recessive deafness 2 (NSRD2). DFNB2 is a form of sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DISEASE: Defects in MYO7A are the cause of deafness autosomal dominant type 11 (DFNA11) [MIM:601317]. DISEASE: Note=Defects in MYO7A may be a cause of Leber congenital amaurosis (LCA), a severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near- absent pupillary responses, photophobia, high hyperopia and keratoconus. SIMILARITY: Contains 2 FERM domains. SIMILARITY: Contains 5 IQ domains. SIMILARITY: Contains 1 myosin head-like domain. SIMILARITY: Contains 2 MyTH4 domains. SIMILARITY: Contains 1 SH3 domain. CAUTION: Represents a unconventional myosin. This protein should not be confused with the conventional myosin-7 (MYH7). WEB RESOURCE: Name=Hereditary hearing loss homepage; Note=Gene page; URL="http://webhost.ua.ac.be/hhh/"; WEB RESOURCE: Name=Mutations of the MYO7A gene; Note=Retina International's Scientific Newsletter; URL="http://www.retina-international.org/files/sci-news/myomut.htm"; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/MYO7A";
hearing loss, sensorineural nonsyndromic Hu, P. et al. 2004, [Mutation screening in selected exons of myosin 7a gene in prelingual non-syndromic hearing impairment patients], Zhonghua er bi yan hou ke za zhi. 2004 Sep;39(9):538-42.
[PubMed 15606003]
The Arg206Gln mutation in exon 7 of myosin 7a is possibly a novel mutation to cause prelingual nonsyndromic hearing impairment. Our results provide the evidence that exon 7 of Myosin 7a is a mutational hotspot region in genetic deafness.
Usher syndrome Ouyang, X. M. et al. 2005, Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population, Human genetics. 2005 Mar;116(4):292-9.
[PubMed 15660226]
The present study suggests that mutations in MYO7A and CDH23 are the two major components of causes for USH1, while PCDH15, USH1C, and SANS are less frequent causes.
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
Pfam Domains: PF00063 - Myosin head (motor domain) PF00373 - FERM central domain PF00612 - IQ calmodulin-binding motif PF00784 - MyTH4 domain
SCOP Domains: 47031 - Second domain of FERM 50044 - SH3-domain 52540 - P-loop containing nucleoside triphosphate hydrolases
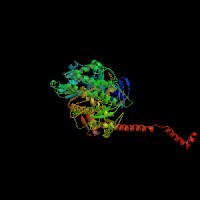
ModBase Predicted Comparative 3D Structure on Q13402
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 US Server
US Server