Description: Homo sapiens all-trans retinoic acid-induced differentiation factor (ATRAID), transcript variant 2, mRNA. RefSeq Summary (NM_001170795): This gene is thought to be involved in apoptosis, and may also be involved in hematopoietic development and differentiation. The use of alternative splice sites and promotors result in multiple transcript variants encoding different isoforms.[provided by RefSeq, Dec 2009]. Sequence Note:. Transcript (Including UTRs) Position: hg19 chr2:27,434,899-27,440,046 Size: 5,148 Total Exon Count: 7 Strand: + Coding Region Position: hg19 chr2:27,435,072-27,439,816 Size: 4,745 Coding Exon Count: 7
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
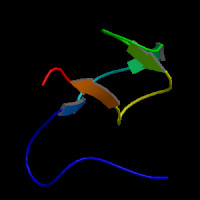
Pfam Domains: PF12661 - Human growth factor-like EGF
ModBase Predicted Comparative 3D Structure on Q6UW56
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Biological Process: GO:0010468 regulation of gene expression GO:0030154 cell differentiation GO:0030501 positive regulation of bone mineralization GO:0033689 negative regulation of osteoblast proliferation GO:0045669 positive regulation of osteoblast differentiation GO:1903363 negative regulation of cellular protein catabolic process
 US Server
US Server